
Cada 21 de junio, el mundo se detiene un momento para reconocer a quienes conviven con la esclerosis lateral amiotrófica. Esa fecha fue elegida en el VI Encuentro de la Alianza Internacional de las Asociaciones ELA, celebrado en 1996 en Chicago, Illinois, con un propósito claro: visibilizar la enfermedad, exigir más investigación sobre sus causas y reclamar tratamientos que mejoren la calidad de vida y la sobrevida de los pacientes. Como futuros profesionales de la salud, entender la ELA en profundidad es un paso concreto en esa dirección.
| INCIDENCIA ~2 por cada 100.000 personas | INICIO HABITUAL A partir de la 5.ª década de vida | FORMA FAMILIAR Hasta el 20% de los casos | ELA BULBAR Mortalidad >50% a los 2 años |
¿Qué es la ELA?
La esclerosis lateral amiotrófica es un trastorno neurodegenerativo progresivo que afecta tanto a las motoneuronas superiores de la corteza cerebral como a las motoneuronas inferiores del tronco del encéfalo y la médula espinal. A medida que estas neuronas mueren, los músculos que innervaban se desnervAN y se atrofian, produciendo una debilidad que avanza de forma inexorable.
Lo que hace a la ELA especialmente compleja es su solapamiento con la demencia frontotemporal (DLFT): no son enfermedades completamente separadas, sino que cada vez se las comprende más como variaciones dentro de un mismo espectro patológico compartido.
Subtipos clínicos
| Subtipo | Motoneurona afectada | Características principales |
| ELA clásica | Superior + inferior | Combinación de espasticidad, debilidad y fasciculaciones; la presentación más frecuente |
| Atrofia muscular progresiva | Inferior predominante | Atrofia y fasciculaciones sin hiperreflexia inicial |
| Esclerosis lateral primaria | Superior predominante | Espasticidad marcada, evolución más lenta |
| Parálisis bulbar progresiva | Núcleos craneales del tronco | Alteraciones tempranas del habla y la deglución; peor pronóstico, >50% mortalidad a 2 años |
Patogenia: proteínas mal plegadas y ARN desregulado
La ELA, tanto en su forma esporádica como familiar, se caracteriza por la acumulación de proteínas tóxicas en las motoneuronas. Este proceso activa vías de estrés celular que terminan desencadenando la apoptosis neuronal. Se han identificado más de 24 loci genéticos causantes de la forma familiar, prácticamente todos con herencia autosómica dominante.
Los genes protagonistas
Las mutaciones más estudiadas convergen en dos mecanismos: agregación proteica anómala y procesamiento aberrante del ARN.
| SOD1 ~20% familiar · Chr. 21 · Ganancia de función tóxica | C9orf72 ~40% familiar · Expansión hexanucleotídica · ELA + DLFT | TARDBP TDP-43 · Proteína de unión a ARN · Inclusiones citoplásmicas | FUS Proteína de gránulos de estrés · Procesamiento de ARN | SQSTM1/p62 Secuestosoma · Autofagia selectiva |
Las inclusiones TDP-43 positivas son el hallazgo neuropatológico más característico: están presentes en la ELA esporádica y en los casos familiares por mutaciones en C9orf72 y TARDBP, pero no en los casos por mutaciones en SOD1 o FUS. Esta distinción tiene relevancia para la clasificación y la investigación de biomarcadores.
Clínica: cómo se presenta y cómo progresa
Los primeros síntomas suelen ser sutiles: debilidad asimétrica de las manos —el paciente refiere que se le caen objetos—, calambres y espasticidad en extremidades superiores e inferiores. Con el tiempo, el cuadro se generaliza y la afectación bulbar cobra protagonismo.
Signos y síntomas por fase

Diagnóstico
No existe un test único que confirme el diagnóstico. Los criterios requieren evidencia de afectación simultánea de motoneurona superior e inferior en distintos territorios corporales (bulbar, cervical, torácico, lumbar), descartando condiciones que la puedan simular como la miastenia gravis, lesiones compresivas medulares o enfermedades autoinmunes.
Los estudios de conducción nerviosa y la electromiografía de aguja son pilares diagnósticos: en la ELA se detectan fibrilaciones y ondas positivas agudas —reflejando la denervación de fibras musculares en reposo—, junto con conducción nerviosa enlentecida. La creatina quinasa (CK) suele estar elevada por la atrofia muscular.
Tratamiento farmacológico: frenando la progresión
Actualmente no existe cura para la ELA. Sin embargo, hay varios fármacos aprobados que modifican el curso de la enfermedad, retrasando la progresión y mejorando la supervivencia en fases tempranas.

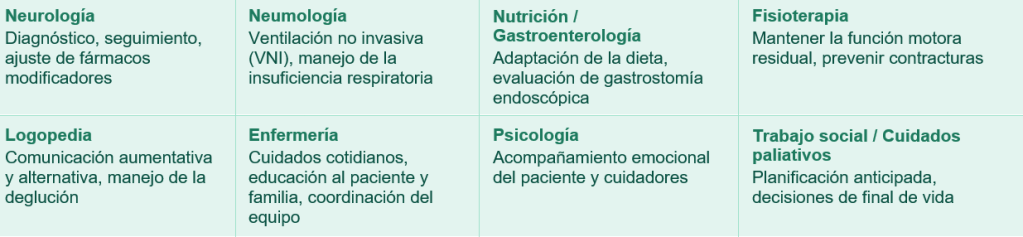
El abordaje interdisciplinario: la clave del acompañamiento
Dado que no existe cura definitiva, la calidad de vida del paciente depende en gran medida de la coordinación entre distintos profesionales. La combinación de estrategias farmacológicas y no farmacológicas es la mejor aproximación disponible hoy.
Aprende ELA —y toda la neurología— con las herramientas que marcan la diferencia
Comprender la ELA a fondo requiere integrar patogenia molecular, fisiopatología clínica y razonamiento clínico. Así es exactamente como están diseñadas las plataformas de Elsevier para estudiantes.
Osmosis: Visualiza lo que los textos no logran explicar
Los vídeos tipo pizarra de Osmosis desglosan la fisiopatología de la ELA —degeneración de motoneuronas, mecanismos de agregación proteica, progresión bulbar— en minutos, sin sobrecarga de información. Ideales para fijar conceptos antes de profundizar con lectura.
Úsalo para: Entender el «por qué» detrás de cada síntoma. Repasa el vídeo de ELA antes de leer el capítulo y verás cuánto más rápido conectas los conceptos.

ClinicalKey Student: la referencia que respalda tu razonamiento clínico
Busca imágenes de alta resolución, crea presentaciones con fuentes citadas automáticamente y practica con preguntas de examen.
Úsalo para: profundizar en los hallazgos morfológicos (inclusiones TDP-43, atrofia de raíces anteriores) y entender la correlación anatomoclínica que el vídeo solo esboza.
Acceder a ClinicalKey Student →
Consejo de estudio combinado
Primero Osmosis, luego ClinicalKey Student. Visualiza el vídeo de ELA para construir el esquema conceptual (motoneuronas, genes, síntomas por etapa). Después profundiza en ClinicalKey Student a través de la última edición de la obra de referencia Robbins, Cotran y Kumar: Patología estructural y funcional para los mecanismos moleculares exactos, los hallazgos neuropatológicos y la correlación clínica. Por último, refuerza con preguntas de examen en ClinicalKey Student. Este flujo —visual → lectura profunda → práctica— es el que mejor consolida la memoria a largo plazo.
{kind=link}
Día Mundial de la ELA: visibilidad que salva vidas
La conmemoración del 21 de junio nació de la convicción de que sin visibilidad no hay financiamiento, y sin financiamiento no hay investigación. Desde 1996, este día ha contribuido a que la ELA deje de ser una «enfermedad huérfana» ignorada por la agenda pública. Cada nuevo fármaco aprobado —desde el riluzol hasta el tofersen— es fruto directo de esa presión sostenida por pacientes, familias y profesionales.
Como estudiante de ciencias de la salud, tu rol en este día no es solo académico: es político y humano. Comprender la ELA en profundidad te prepara para ser el profesional que el sistema de salud necesita junto a estos pacientes.
La ELA nos recuerda que la medicina no siempre puede curar, pero siempre puede acompañar. El abordaje interdisciplinario es hoy la mejor herramienta que tenemos para garantizar que esa compañía sea digna, informada y compasiva.
Este 21 de junio, súmate a la visibilidad: habla de la ELA, comparte lo que aprendes, y lleva esa comprensión a la cabecera del paciente.
Contenido basado en:
Deja una respuesta